Inside of the Rare Disease - Tyrosinemia
Published on 28 Sep, 2020
Historically, rare diseases are neglected primarily because of lack of disease awareness and diagnosis, limited patient population, and sparse epidemiological data. However, in recent past, rare diseases are witnessing intense R&D activity as companies are leveraging drugs for rare diseases due to less regulatory hurdles, shorter time-to-market, exorbitant cost of drugs developed, and extended market exclusivity. In this article, we provide insights on unmet medical needs, patient journey, treatment algorithm, and future outlook for tyrosinemia - a rare disease affecting 1 in 100,000 people.
The market for tyrosinemia is currently valued at USD 99 Mn, having recorded a compound annual growth rate (CAGR) of ~1% from 2013 to 2018. The subdued growth can be attributed to low rate of diagnosis of the disease and limited options for treatment.
Until recently, Nitisinone [(2-(2-nitro-4-trifluoromethylbenzoyl) cyclohexane 1-, 3-dione, NTBC)] was the only available therapeutic treatment option for those afflicted by tyrosinemia. Prior to this, low protein (tyrosine and phenylalanine) diet was the single recourse available to halt its progress. Two drugs in the market, both with Nitisinone as an active pharmaceutical ingredient (API), were Orfadin (capsule/suspension) by Swedish Orphan Biovitrum International AB and Nityr (tablet) from Cycle Pharmaceuticals.
In May 1995, the Office for Orphan Product Development gave NTBC the Orphan Drug Designation. In January 2002, NTBC received the US Food and Drug Administration (FDA)’s approval. In 2005, European Medicine Agency (EMA) approved Nitisinone under exceptional circumstances.
FDA evaluated NTBC in an open-label study involving around 180 pediatric patients from around 25 countries at 87 sites. The study revealed a four-year survival rate of around 88% with children less than two years old. Nitisinone is generally prescribed at 1.0 mg/kg/day, although individual requirements may vary. Dosage should be adjusted to maintain Nitisinone levels in the blood at 40–60 µmol/L.
In 2017, FDA approved Nityr (Nitisinone tablets) for the treatment of hereditary tyrosinemia type I (HT1), following the expiry of SOBI’s Orfadin. Nityr tablets are the bioequivalent of Orfadin (Nitisinone) capsules. The tablets can be administered with or without food and are available in strengths of 2 mg, 5 mg, and 10 mg. However, Nityr tablets do not need refrigeration and can easily disintegrate in water; therefore, they can be easily administered to pediatric patients.
Disanit, manufactured by Italy-based Dipharma, is an improved generic version of Orfadin. The drug is currently being evaluated for marketing authorization in Europe for the treatment of HT1. Apart from this single generic drug, there are no other new drugs in the pipeline.
What is the pathophysiology of tyrosinemia?
The fumarylacetoacetate hydrolase (FAH) gene produces the FAH enzyme. Deficiency of FAH leads to an increase in fumarylacetoacetate and tyrosine and their metabolites in the kidneys, liver, and central nervous system (CNS). A defect in FAH gene results in tyrosinemia, a rare disease.
There are three forms: tyrosinemia type I (HT1), type II (HT2), and type III (HT3). HT1 affects both genders equally, whereas types II and III are comparatively less common.
HT1 is an autosomal recessive genetic disorder that leads to the deficiency of the FAH enzyme, thereby affecting the tyrosine breakdown. The various symptoms associated with tyrosinemia include failure to gain weight, developmental abnormality, fever, diarrhea, vomiting, hepatomegaly (an abnormally large liver), and jaundice-like symptoms (yellowing of the skin and eyes).
The disorder, if left untreated, can lead to further complications such as liver disease, renal tubular dysfunction, cirrhosis, and hepatocarcinoma. The risk of hepatocellular carcinoma (HCC) is estimated to be 18–37% in untreated tyrosinemia patients.
Notably, around 10% of newborns have temporarily elevated levels of tyrosine (transient tyrosinemia). This is not a genetic condition and usually occurs due to deficiency of vitamin C or liver enzymes dysfunction primarily on account of premature birth.
How prevalent is tyrosinemia?
Given the inconsistency in clinical presentation, it is estimated that less than 50% of afflicted individuals get diagnosed with tyrosinemia.
Around 1 individual in 100,000 people is affected with HT1 worldwide. It is estimated that 70,000 children across the globe are affected with tyrosinemia.
The highest number of cases is registered in Norway, Sweden, Finland, and Quebec in Canada, mainly owing to the high rate of diagnosis in these countries.
How is the disease diagnosed?
The symptoms of tyrosinemia in infants are low physical growth and hepatomegaly (an enlarged liver). Presence of tyrosine metabolites and succinylacetone in the urine confirms tyrosinemia. Diminished FAH activity in liver tissue or cultured fibroblasts is also an indicator; however, the test is not available easily.
Tandem mass spectroscopy is one of the ways employed to measure succinylacetone in newborn blood spot screening. DNA analysis is recommended to detect tyrosinemia for families where specific gene-causing mutation has been identified.
Next-generation DNA sequencing techniques such as exome sequencing and whole genome sequencing (WGS) assist in detecting mutations responsible for the disease.
Baseline tests such as blood/plasma, blood gases, liver function tests (bilirubin, aspartate and alanine aminotransferase), glucose and ammonia, full blood count, α-fetoprotein (AFP); ultrasound of liver and kidney; and MRI for nodules are the other investigations to confirm tyrosinemia.
Prenatal diagnosis, entailing evaluation of succinylacetone and DNA analysis in amniotic fluid, can be performed as well. Molecular genetic testing for FAH gene mutations is a confirmatory test for tyrosinemia diagnosis.
What are the unmet medical needs?
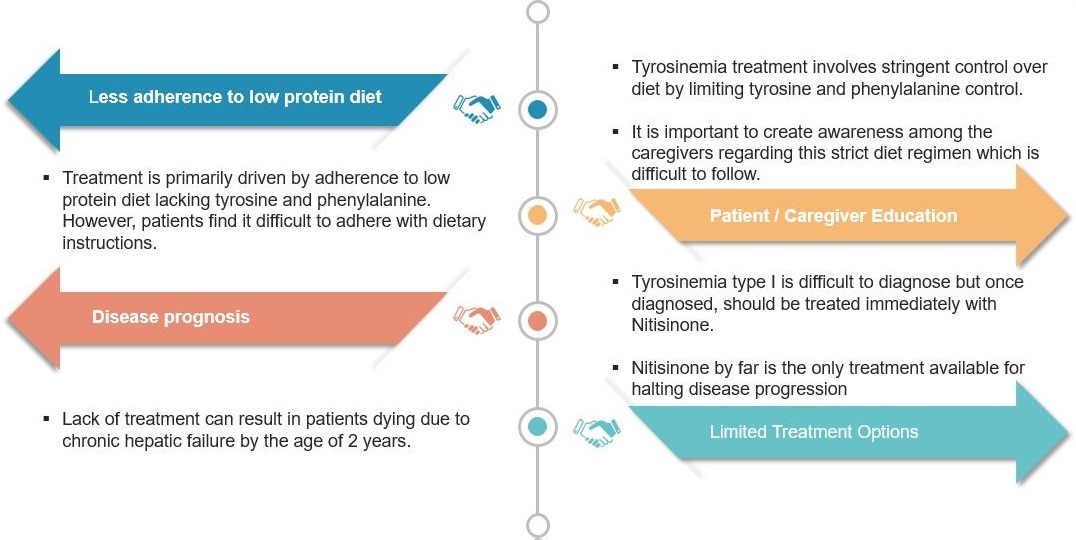
If untreated, HT1 may lead to chronic liver failure and hepatic cancer. Infants suffering from this fatal disease will not survive beyond two years in the absence of timely treatment.
The main issues with HT1 are that it gets rarely detected and options for treatment are few. Moreover, upon diagnosis, treatment should start immediately involving administering of Nitisinone and a strict diet. While the diet is difficult to follow, it is essential to improve the patient’s condition.
Exhibit I – Unmet Medical Needs
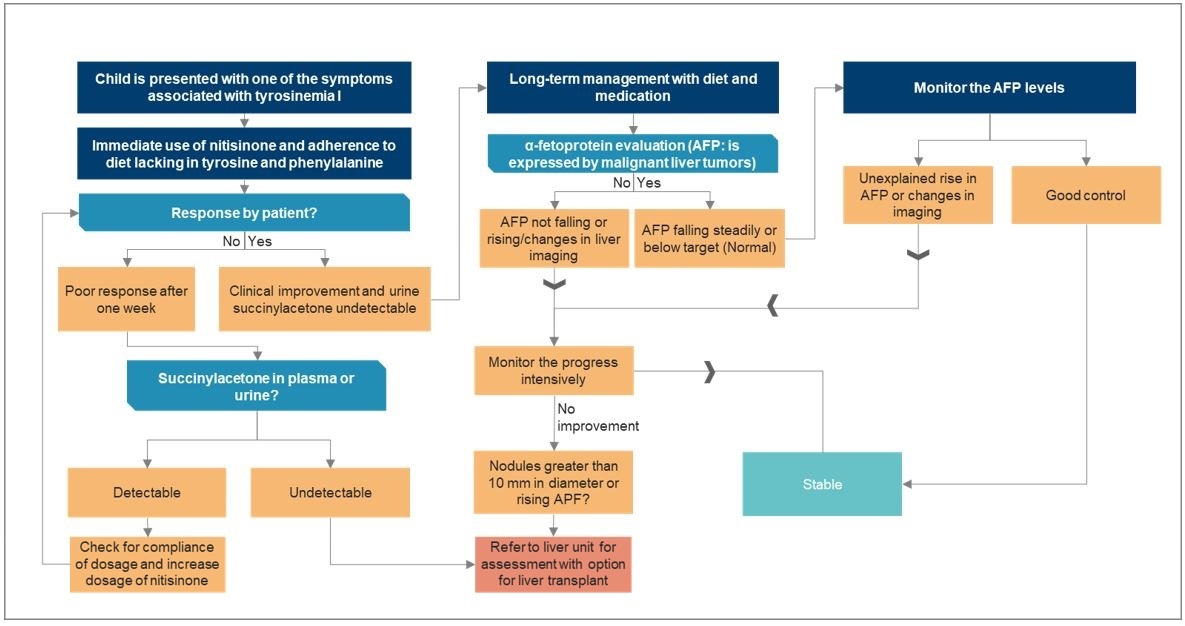
What is the patient journey and treatment algorithm of tyrosinemia?
Patients (newborns) with tyrosinemia experience hepatomegaly, splenomegaly, cirrhosis, liver failure, tubulopathy, nephromegaly, Franconia syndrome, seizures, and low growth rate.
Typical age of patients at the clinical onset of tyrosinemia is around 9 months. The mean age of diagnosis is reported to be ~16.3 months. Patients receiving supportive care or nutritional treatment have a three-year survival rate of 10%. Patients undergoing liver transplantation have a six-year survival rate of 60%.
The leading causes of death among these patients are fulminant liver failure, porphyria-like neurologic crisis, and metastatic hepatocellular carcinoma.
Exhibit II – Patient Journey and Treatment Algorithm – Tyrosinemia Type I
What are the available treatment options?
As mentioned earlier, Nitisinone was the only approved and available treatment for HT1. It was discovered incidentally, and is a byproduct of agrochemistry. Orfadin, a synthetic reversible inhibitor of 4 hydroxyphenylpyruvate dioxygenase, is prescribed, along with dietary restrictions of tyrosine and phenylalanine.
Treatment with Nitisinone and a low-tyrosine diet should commence on priority after diagnosis. Initiation of Nitisinone therapy in the initial period is warranted to prevent HCC, liver and kidney dysfunction, rickets, and neurological disorders.
To prevent tyrosine levels from shooting up, a low-protein diet, along with amino acid mixtures devoid of tyrosine and phenylalanine, is recommended. The plasma tyrosine levels should be kept below 400 µM. Nitisinone does not have major side-effects; eye symptoms may appear but are reversible.
Liver transplantation is the last resort to tackle tyrosinemia. It is recommended for children who have end-stage liver failure at initial diagnosis and are unresponsive to Nitisinone therapy, or who have documented evidence of malignant changes in hepatic tissue.
Transplant recipients require long-term immunosuppression. Mortality associated with liver transplantation in young children is approximately 10%. Transplant recipients can also benefit from low-dose (0.1mg/kg/day) Nitisinone therapy to prevent ongoing renal tubular and glomerular dysfunction resulting from high levels of succinylacetone in the plasma and urine.
Physicians experienced in treating HT1 are deemed legitimate to prescribe these medicines as the dosage should be decided based on the patient’s weight and biochemical tests. Nutritionist play a key role in managing children with inborn errors of metabolism who require a low-protein diet. To ensure the right dose for the patient is maintained, blood test should be performed routinely.
What is the outlook for tyrosinemia management?
-
The market for tyrosinemia is expected to remain stagnant, declining marginally to USD 98 Mn by 2021 primarily due to:
- Low disease diagnosis rate
- Limited patient pool
- Single treatment option
- Expensive treatment [the cost of Nitisinone per person per year (PPPY) is estimated at USD 51,493 ]
- To improve the quality of life for patients, newborn screening programs, in combination with administration of orphan drugs, proper monitoring, genetic counselling, and following clinical practice guidelines, are indispensable. In the light of the present scenario, there is significant scope for evaluating and commercializing better treatment options for curing tyrosinemia.

Vasuda Venkitakrishnan
